 
| J・クレイグ・ベンター |
|
|
|
 |
 |
|

[figure 1]

[figure 2]

[figure 3]
[figure 4]

[figure 5]

[figure 6]

[figure 7]
|
Thank you very much, Dr. Matsubara, Dr. Takeda, Members of the Jury, Fellow Laureates, Ladies and Gentlemen. [Figure 1] It's indeed a pleasure to be here to talk to you about sequencing the human genome, how we got there and what some of the implications are.
I started my career as a biochemist. I spent ten years trying to purify just one protein, but it became clear that the techniques for protein sequencing at the time were not sufficient to obtain the sequence of a protein in an easy fashion unless you had a very substantial amount of protein. The major revolution that started was to be able to clone a cDNA, sequence the DNA, and then determine the protein structure from the DNA. That's what allowed major advances in the protein world. And as you heard, my colleague, Dr. Hunkapiller, developed much of the technology that allowed that revolution to happen. I am going to show you now, as we go into genomics, that the world is going to turn a complete 360 degrees, allowing us now to go back and do in the protein world what protein biochemists wanted to do all along -- enabled for the first time by the sequencing of the human genome.
I was very much influenced by a key paper published by Dr. Hunkapiller and Lee Hood and colleagues in 1986 in Nature describing the new technique that they had developed. It involved modifying Sanger sequencing with fluorescent dyes, allowing, as Mike (Hunkapiller) showed, the information to be read out into a computer. You will see from that point on, for the last fifteen years, there has been a tremendous parallelism between Mike's career and successes and my career and successes, based independently on the approaches that we have come up with that ended up helping the other quite substantially.
In early 1987, my lab became the first test site for that new sequencing machine that Mike's team developed, and we sequenced the first genes and published them in our paper in the Proceedings of the National Academy of Science (PNAS) in 1987. We started the first test projects at NIH to sequence parts of the human genome to see if, in fact, it was feasible. I remember clearly some discussions that Mike and Lee Hood and I had in 1987 where we talked about the human genome one day being sequenced in a large factory full of these machines. I don't think either of us realized that it would take fifteen years to get back around to that idea. The new technology allowed us to do things that we never envisioned before. Even just a few years ago we didn't think it was possible to accomplish what we have just accomplished recently. I was in Japan on a speaking tour, along with others, with Applied Biosystems when I entered into a series of discussions with Dr. Matsubara and his colleagues. Dr. Matsubara, as many of you know, was an early proponent of cDNA sequencing in the human genome. On my flight back I thought of a new approach to apply this with, and this led to the expressed sequence tag (EST) method. At almost every level, from the earliest conceptual discussions to the Hitachi contribution to the DNA sequencer, there has been a tremendous Japanese influence throughout the genome effort.
[Figure 2] In 1991, we published this paper in Science describing the EST method that allowed us to discover genes very rapidly by sequencing large number of cDNAs. However, it was the challenge of dealing with all this data that led to the discoveries that took us to the next level. When we formed The Institute for Genomic Research (TIGR) in 1992, we had the largest sequencing lab in the world at that time. In the first year we sequenced approximately 100,000 cDNA clones; that seemed like an absolute breakthrough. Now, that's about half a day's work at Celera, but it took a whole year with the largest factory at the time to do this work. Our challenge was putting all the data back together again to understand how all these sequences were related to one another and maybe how many human genes there were. We hired a mathematician, Granger Sutton, to develop a new algorithm with the team at TIGR that is now called the "TIGR assembler." My colleagues and I, including Ham (Hamilton) Smith, the Nobel Laureate who discovered restriction nucleases, were sitting around discussing how we could apply this new algorithm to genomics, and we decided to see if we could sequence a single genome. We wrote a grant application in 1994 and submitted it to the National Institutes of Health (NIH), but we were told that what we were doing was impossible and wouldn't work. [Figure 3] So, we used the endowment money that we had at TIGR, and we ended up sequencing the first genome of a free-living organism in history in 1995.
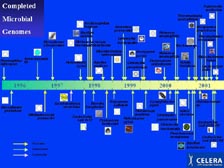
[Figure 4] Genomics has a very short history. The first sequenced genome of a self-replicating organism was only 7 years ago. The method allowed very rapid development, and over 90 percent of the genome now sequenced in the world have been sequenced with our whole genome shotgun method. An important note for Japan is that if it had not been for TIGR, the first genome sequenced in history would have been from Japan. Therefore, I am indeed honored that you will have me back here. The first one was Haemophilus influenzae, followed by Mycoplasma genitalium. However, Synechocystis sp., the first blue-green algae, was sequenced here, in Japan, followed by the first Archaea at TIGR. As Mike showed, the number of genomes being sequenced is growing exponentially, even though the original plan from the U.S. government during this entire period was just to have the E. coli genome sequenced -- so the new algorithms and the new technology have allowed the world to change quite dramatically.
We had sequenced a large number of genomes at TIGR, and we felt that these approaches would work for the human genome. In fact, if you go back and read the Haemophilus influenzae paper that we published in Science in 1995, at the end of the paper we said: "This is the method that will be used one day to sequence the human genome." What was lacking was the appropriate technology. We knew the stars were out there, but we didn't have a powerful enough telescope to see them. Therefore, when I got the call from Mike saying that his team had developed this exciting new instrument, it became very clear to us very quickly that we could go much further and see much further. We agreed, after a one-day meeting, to form Celera Genomics to sequence the genome, with money from the former Perkin-Elmer Corporation. [Figure 5] We built this large factory. It's the size of a football field and had 300 of these instruments in it. Even so, this facility is only run by 7 people that work 24 hours a day, 7 days a week. That was in contrast to as many as 10,000 scientists in the public effort to generate similar amounts of data.
[Figure 6] Our challenge was handling all the information. At the time, there were no computers that were available for this. We worked with Compaq computer to build a one and a half teraflop computer, which means it can do roughly 1.5 trillion calculations per second. Even so, it took weeks to assemble the human genome using this computer. Right now we have over 100 terabytes of spinning disk, and you will see as I go forward how that is going to grow by orders of magnitude. Our biggest limitation in genomics right now is the size of the computations that can be done, not the amount of DNA that can be sequenced.
[Figure 7] We thought it was important to do a test project priort to sequencing the human genome. We chose the fruit fly Drosophila Melanogaster. The critics who said that we could not sequence the Haemophilus influenzae genome were even more critical when we decided to go up two orders of magnitude in scale and try to sequence all of the Drosophila genome in less than one year. I asked Gerry Rubin, the Head of the Drosophila Genome Group to collaborate with us, and he accepted immediately. We had a very rapid process with which to proceed. |
|
|

