 
| マイケル・W・ハンカピラー |
|
|
|
 |
 |
|

[figure 1]
[figure 2]

[figure 3]
[figure 4]

[figure 5]
[figure 6] |
Thank you for
that kind introduction, Dr. Matsubara. I would also like to extend
my thanks to Dr. Takeda and the Directors and staff of the Takeda
Foundation for putting on this wonderful symposium and inviting
us to Tokyo. [figure 1] To give you a sense of the passion that
Craig (Venter) and I have developed over the last several years
for the life sciences research field, I will tell you that we think
the benefits to mankind that can come from leading edge research
to understand fundamental human biology, carried out literally around
the world, are enormous. From this understanding, we will be able
to develop the kind of insights and tools that will allow people
to have improved lives and longevity.
I will begin my comments by saying that when Craig (Venter) was first presented with the technology and the idea of sequencing the human genome, he was shocked. However, we looked at the possibility together and decided maybe it was doable. We may have been crazy at the time, but with Craig's hard work and the team he was able to put together, sequencing the human genome was, in fact, a doable project.
In my remarks, I am going to try to give you not just a sense of how the development of the technology to carry out the sequencing of the human genome occurred and what was involved, but also a sense of where people will go from here. I will present the future development of the technology in terms of what is needed to help further the understanding of this blueprint of life, i.e. the genetic code.
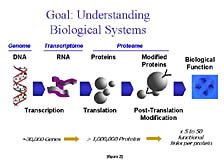
The goal of the researcher -- to understand how biological systems work at the fundamental biochemical level -- is one that Applied Biosystems supports with its technology. [figure 2] In order to achieve this level of understanding, researchers need to know the structure and the function of several key, complex, biological molecules. The most important of these, from an information/content perspective, are nucleic acids, more commonly referred to as DNA and RNA. Each has a separate job. DNA's role is to act as a fixed repository of information that can be transferred from one generation to another. RNA's task is to carry out the DNA's instructions for particular biological processes at given stages in a cell's life. As Craig has discovered, and as the Human Genome Project has discovered, there are only about 30,000 different genes that are the core building blocks of DNA structure. These genes give rise to the transmittance and the use of the genetically stored information.
The information that's housed in nucleic acids is translated into activity when a cell wants to put it to use in the form of a different kind of complex, biological molecule called a "protein." Proteins are then sometimes broken down into smaller groups of peptides and can be further modified with the addition of other kinds of chemical groups to carry out the specific function for which the protein is intended. While there are only about 30,000 genes in the human genome, there are upwards of a million or so different proteins, peptides, and modified forms of these compounds that can be active in a cell at one time or another during the course of an organism's life.
Proteins function not only as individual molecules to carry out a specific chemical reaction or perform a specific structural task, but they also form networks in which they work together in different ways, at different times, under different conditions in order to carry out much more complicated tasks. Therefore, unraveling biological function requires a lot of information, both about the specific structural components of the individual molecules that make up these nucleic acids, or protein networks, and about the three-dimensional structures that give rise to a lot of the functional characteristics that are imparted by that structure contained within them.
When we looked at the problem of how one looked at these processes from a genome-wide or proteome-wide, or cell-wide level, several years ago at Caltech, it was pretty clear that biology, at that time, was a manually driven process. Automation had made inroads into the drive to improve chemistry, but hadn't hit biology as much as we thought would be necessary in order to carry out large-scale studies. Automation was needed to begin to get a look at not just the very tiny pieces of living cells (e.g., one protein, one gene, or one cell), but to study the entire component of those molecules. [figure 3] So, while at Caltech, Lee Hood and myself, and others, proposed the concept of transforming the way biology is done at the basic research level to take advantage of modern tools for doing large-scale structural and functional analysis of these molecules.
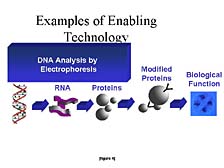
I would like to give you some insight into three classes of these tools, with a good part of the focus illustrated in this slide, and to talk about the tools necessary for doing structural characterization of genes and related nucleic acid fragments. [figure 4] The basic tool, as was mentioned by Dr. Matsubara, is one known as "DNA electrophoresis," which separates molecules on the basis of their size. [figure 5] It was the fundamental understanding of the structure of DNA, by Francis Crick and Jim Watson in the early 1950's, that laid the groundwork for this kind of tool to be widely used in the study of nucleic acids. Contained within nucleic acids, in the order of the components that make up one gene versus another and one part of the chromosome versus another, is the simple, but elegant, ability to carry the information necessary for their own replication. It was this realization and the development of tools by Fred Sanger, at Cambridge, and others in the mid-1970's that gave rise to the use of DNA electrophoresis as a tool for studying the basic ordering of the components within nucleic acids. They each have only four constituents that make them up. It's the order and the number of these constituents that give rise to the differences between one gene and another.
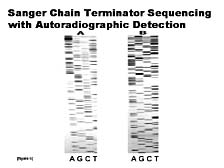
The concept, here, was to use a lot of the fundamental biochemistry that's involved in normal cell replication of nucleic acids as a means of identifying and elucidating the structure of these molecules in terms of their basic sequence. [figure 6] What was generated, and what's called the "Sanger chain terminating sequencing methodology," is a series of fragments of DNA, with the original implementation carried out with four separate reactions, one for each of the four types of nucleic acids that can make up DNA. By separating the fragments by size using electrophoresis, and realizing that the smaller pieces have an order relative to the larger ones, and that this order is related to the sequence, you can determine the sequence of the DNA. The shortest piece would be an A, the next piece might be a C, the next one an A, a T, and so forth. By manually going through and reading the ladder from top to bottom you could determine the sequence. The beauty of this technique was that it worked; the drawback was that it required somebody to sit there, look at this picture and the separated ladders of DNA, and manually interpret the sequence. You can see, without much knowledge of the details, that the signals seen in the form of these bands are not uniform. If there are any anomalies, in terms of how fast the bands run in part of the gel versus another, it might make the sequence complicated to read. In fact, this was often the case. Thus, the Sanger method was a fairly error-prone process in the beginning.
|
|
|

