| J. Craig Venter |
 |
|
|
 |
 |

|

|
J. Craig Venter |
|
Celera Genomics |
|
|
 |
|
[figure 8]
[figure 9]
[figure 10]

[figure 11]
[figure 12]
[figure 13]
[figure 14]

[figure 15]

[figure 16]

[figure 17]

[figure 18]
|
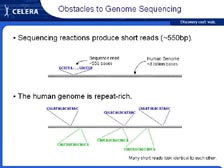
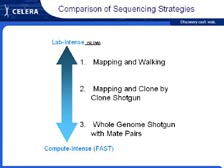
[Figure 8] However, let me tell you a little bit about the challenges with genome sequencing -- why is it so difficult and why did we needed new methods. The instruments that Mike told you about give us somewhere between 500 and 700 letters of genetic code at a time. So the challenge is how do you sequence something that's millions or even billions of letters of genetic code long when you can only get 500 to 700 letters at a time? In addition, every genome that we have worked on is absolutely full of complex repeats; some of these are short and some of these are quite long. Lots of methods were discussed. [Figure 9] Initially mapping and walking were discussed. By this method you sequence 500 letters of genetic code, then you make a new primer that allows you to sequence the next 500 letters in order. It was calculated that it could take over 100 years to sequence the human genome using that approach.
Sequencing was very much influenced by Fred Sanger and his accomplishments. He sequenced the largest viral genome that was done as a single project, bacteriophage lambda, which was only 47,000 letters long. That was a tremendous accomplishment in the early 1980's, but it influenced the thinking going forward that nothing larger than lambda could be sequenced. Therefore, when people talked about sequencing large genomes, they had to break these down into thousands and thousands of smaller projects. So, the first phase people talked about was mapping, where these clones were made and then lined up in an order along the chromosomes. Once that order was determined, they were then sequenced. We thought that there had to be a single mathematical solution for each of our genomes. Otherwise, life should not be possible. We would not be able to have children in a reasonable fashion. There would be just too many mutations and changes. It was with the Haemophilus influenzae genome that we developed the whole genome shotgun method that we thought would work with the human genome.
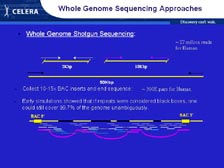
[Figure 10] When most laboratories around the world were trying to sequence the assembled DNA, they sequenced just one stretch of 500 letters and then tried to overlap it into a computer with an exact match of another stretch of 500 letters. Therefore, you could build only small local structures until you had a very substantial amount of data. The key strategy that we developed first with H. influenzae -- which was absolutely essential for Drosophila and human and mouse -- is that we sequenced both ends of clones of different sizes. [Figure 11] This created scaffolds, the same approach that you use for building large buildings and long bridges. We had pieces that were 2,000 letters long, pieces that were 10,000 letters long, a new approach to develop pieces that were 50,000 letters long, as well as pieces that were 150,000 letters long. We got 500 to 600 letters of genetic code from both ends of each of those clones. Our computer algorithms were able to track those very carefully and keep them together. Therefore, if we knew where one piece was, we knew the other end was either 2,000 letters away, 10,000, 50,000 or 150,000. I think you can see how, by overlapping tens of millions of these pieces, you could build a large letter structure very quickly. The people in the public genome projects that were just looking at single reads thought it would be impossible to assemble these large genomes because of all the repeats and because you could only get these small structures put together. We totally overcame these obstacles with the paired end-sequencing method. This was as important as the algorithms were to developing the genome.
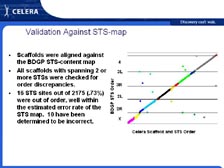
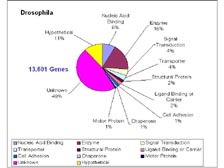
[Figure 12] The other approach that we took involved the fact that many sequences will have multiple sites that they match in the genetic code. Therefore, we only put pieces together if there was a single mathematical solution. If there were two different sites that a sequence could go to, we left it alone. And then if we required two of these paired end links to go between these smaller assemblies, there was less than one chance in 1015 (ten to the fifteenth) of making an error doing this. The structures went together very rapidly and became extremely accurate, with only small holes in the genome where repeat sequences would go. [Figure 13] When we did this in four months with the Drosophila genome, and we assembled it and compared it to all the mapping data, there were only 16 sites that didn't agree with our sequence. It was found later that all 16 of those were errors made in the mapping of the genome. Gerry Rubin's group has reported that the Drosophila sequence is the most accurate sequence ever published, with less than one error per 1 million letters of genetic code. We had a team that annotated the genome quickly, and we've found that over half of the genes in Drosophila, despite more than 100 years of study, were new to science. This is what we have seen in virtually every genome sequenced to date, from H. influenzae through to the mouse, creating a tremendous challenge for science. The scientists at TIGR, alone, have published over 100,000 new microbial genes of unknown function that are in GenBank. In the United States, in Japan, and in the rest of the world, we do not have funding mechanisms that will allow scientists to study these unknown genes. You basically have to know the answer before you can get money to study them.
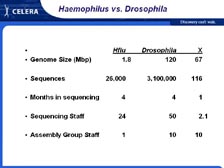
[Figure 14] If we look over just a short period of time -- because in terms of methodology, the two key papers to me are the H. influenzae genome and the Drosophila genome -- the human genome, in terms of sequencing, became totally obvious after Drosophila. However, there was a big difference in just a few years time in terms of what was possible because of the new sequencing technology. Haemophilus influenzae took four months of sequencing. Drosophila took four months of sequencing. After the building and scaling up Celera we were able to sequence 29 million sequences in nine months. Then we turned around and sequenced another 29 million sequences from the mouse genome in only 6 months, showing a continual change in the speed of DNA sequencing.
[Figure 15] As soon as we finished Drosophila, we started sequencing the human genome. We had a number of normal volunteers, at least quasi-normal volunteers, because they were all scientists. [Figure 16] We made sure they had a complete set of chromosomes, and we chose 5 for sequencing: 3 women and 2 men, of self-described ethnicity as African American, Hispanic, Chinese and Caucasian. It took 9 months to sequence the genome, but in that time these 29 million clones covered the genome over 39 times in these paired end scaffolds.
[Figure 17] The first assembly was announced at the White House on June 26th, 2000. I was told that it was the first scientific announcement ever to be made from the White House, and the first ever on live, international television. It was an exciting event for science. Earlier this year (2001), in Science, we published our analysis of the human genome. This and the accompanying paper in Nature by the publicly funded group, we were just informed, are already citation classics. They now hold the world's record for the most citations in the shortest period of time before the year is even completed. I had lots of help. [Figure 18] There are 282 co-authors on this paper. There was a tremendous challenge trying to get the first description of the human genetic code. Knowing that this paper would be looked at for a very long period of time, and scrutinized around the world, we wanted this to be as accurate as possible. Not all of these authors contributed to the writing. Many of them contributed to designing the software or doing the sequencing or helping with different parts of the analysis.
|
|
|