| J. Craig Venter |
 |
|
|
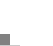 |
 |

|

|
J. Craig Venter |
|
Celera Genomics |
|
|
 |
|
[figure 41]
[figure 42]
[figure 43]
[figure 44]
[figure 45]
[figure 46]

[figure 47]

[figure 48]

[figure 49]

[figure 50]

[figure 51]

[figure 52]
|
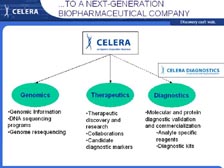
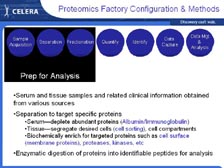
[Figure 41] At Celera, we are now trying to use the genome to come up with new therapies to treat cancer and other diseases. [Figure 42] Using all this information we are taking gene family approaches and disease specific approaches (with the new proteomics pipeline that I will show you) that can lead to new diagnostics, new targets for small molecules and new targets for cellular immuno-therapeutics and monoclonal antibodies. [Figure 43] Over the last 18 months we have built a large proteomics pipeline with all new methodology. We don't have any 2-D gels. We developed new approaches for fractionating tissues, doing cell fractionation into sub-components, and sequencing individual proteins using the new mass spectrometers that Mike detailed in his lecture.
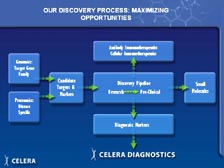
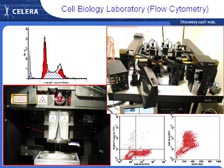
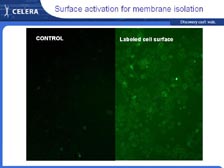
We can now go back to what I was trying to do fifteen years ago, and that is get the sequence of individual proteins very rapidly without having to go to the DNA. With tumor tissue right out of patients, we can use cell sorters that sort 50,000 cells per second, using four different lasers to put the cells in different populations. [Figure 44] Then, using new cell biology techniques, we can fractionate those individual populations of cells. [Figure 45] We have developed a new proprietary technique for obtaining the cell surface proteins -- getting these proteins out and putting them right into the mass spectrometer. In the past, scientists had to have 2-D gels to get individual spots of theoretically pure proteins before they could sequence them in a mass spectrometer. Now with the new mass spectrometers and the complete genome to sort out the data, we can do all this and sort out the information at the end. So, we are trading upfront resolution with low resolution at the beginning to get exquisite high-throughput resolution at the other end.
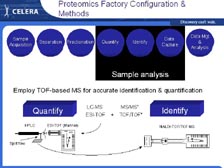
[Figure 46] We have split flow off the high-performance liquid chromatography (HPLC) system, where we use one type of mass spectrometer to get the quantity of proteins in the cells with differential expression (including the ICAT Reagent that Mike talked about) and take the other half of the flow into the new T2 system, the time of flight mass spectrometer that can sequence large numbers of individual proteins. [Figure 47] Therefore, we have a very large mass spectrometry laboratory, the size of our DNA sequencing laboratory, with nine of the TOF-TOF (T2) systems and a very large number of Mariners and Q-Stars for sorting the proteins very quickly.
[Figure 48] We are taking serum directly from patients with different cancers, and we are sequencing the proteins that are expressed, looking for new markers that can be an early diagnostic tool to determine whether you have lung cancer or breast cancer or colon cancer. We are taking the tumor cells through cell separation into the mass spectrometers to find new, differentially expressed proteins. We don't care what their functions are. As long as we know each has a unique differential expression, we can make T-cell vaccines against those proteins. If they are from the cell surface category, which we know the sub-side of the categorization right off the mass spectrometer, they can go until monoclonal antibody program to make an immunotherapeutic or screening with chemistry molecules.
[Figure 49] In all cases, we are trying to revolutionize drug discovery by eliminating the middle steps of identifying targets and then validating them. We go directly to producing the new drug -- whether it's a monoclonal antibody or a small molecule -- and then we go back and validate the target with disease in the end. The advantage with proteomics that we have is the computing power to deconvolute all this information. We are already dealing with terabyte volumes in very short time periods.
[Figure 50] At Celera South San Francisco, we have over 100 medicinal chemists that deal with diverse libraries, now close to a million compounds, in a new state-of-the-art chemistry factory that allows us to do very rapid small molecule identification. We can go right from the mass spectrometers to producing large amounts of proteins and getting the X-ray crystal structure of those proteins. [Figure 51] In the last year, the group did over 140 X-ray crystal structures, many of these with the drug-bound, and designed a number of new protease inhibitors based on knowing the precise protein structure. We are in the process of scaling up this rate of X-ray crystallography, by more than one order of magnitude, to increase the rate of structural base drug design. We can discover new proteins associated with cells, rapidly determine those protein structures and design specific inhibitors to work on those, and then take those drugs back into animals and individuals to understand the disease state.
[Figure 52] Therefore, you can see in a very short period of time, less than 15 years, we have gone from no sequenced genomes to a tremendous amount of genomic data. Right now at Celera we have over 250 terabytes of data, and it's growing exponentially. As we develop our protein analysis, we are already getting into the petabyte data range. We are trying to deconvolute this information and to understand individual variations with disease and complex analysis of proteins. If there are 250,000 proteins in 100 trillion cells, then to understand all the different combinations you have to take 250,000 to the 100 trillion power to have the total number of possible protein-protein interactions. New computing power is going to have to be developed for human biology to be understood and to develop rapidly.
I think I have shown you how, over the last fifteen years, we have gone from spending decades on individual proteins to where people can make those same discoveries in about a 5 second search of our database. The challenge now is integrating biology. We have the first chance ever to have a holistic look at how all our components work together to yield human life and life of other species, and I think this is going to be an extremely exciting era as we go forward. Thank you very much.
MODERATOR: Thank you very much, Dr. Venter, for a wonderful presentation.
|
|
|